
专家解读 | CHO重组表达产品上市申报前遗传稳定性研究要求
2024/12/18

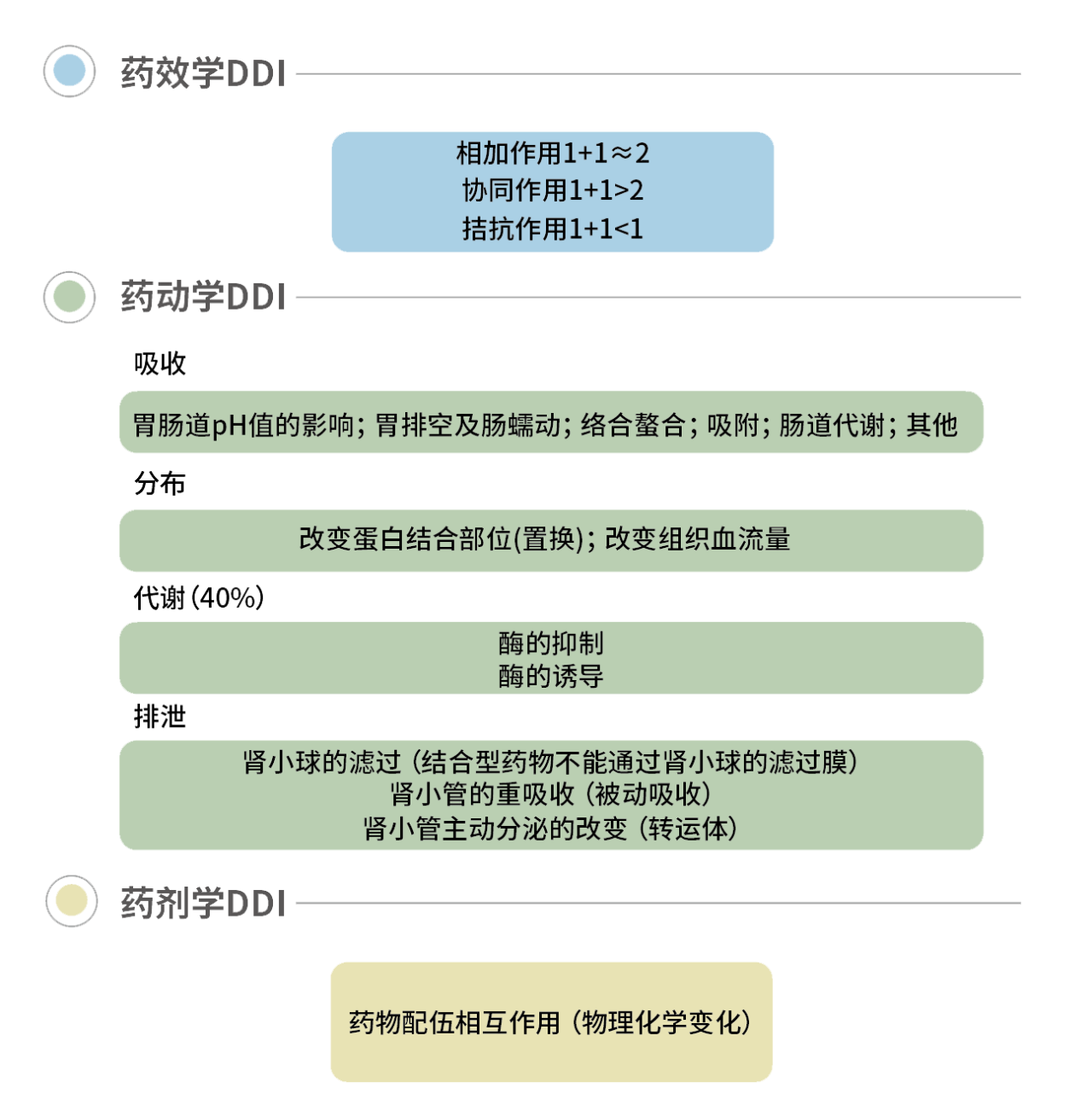
按照发生DDI的原理,可分为:
1
药效学DDI
相加作用1+1≈2
协同作用1+1>2
拮抗作用1+1<1
2
药动学DDI
吸收:胃肠道pH值的影响;胃排空及肠蠕动;络合螯合;吸附;肠内代谢;其他
分布:改变蛋白结合部位(置换);改变组织血流量
代谢(40%):酶的抑制;酶的诱导
排泄:肾小球的滤过(结合型药物不能通过肾小球的滤过膜);肾小管的重吸收(被动吸收);肾小管主动分泌的改变(转运体)
3
药剂学DDI
药物配伍相互作用(物理化学变化)
一、代谢酶介导的DDI研究策略
药物代谢的主要场所是肝脏。肝脏进行生物转化则依赖于微粒体中的多种酶系,其中最重要的是细胞色素P450混合功能氧化酶系统。代谢性相互作用的96%是由P450酶系介导。
正常人肝微粒体10种CYP450含量的构成比
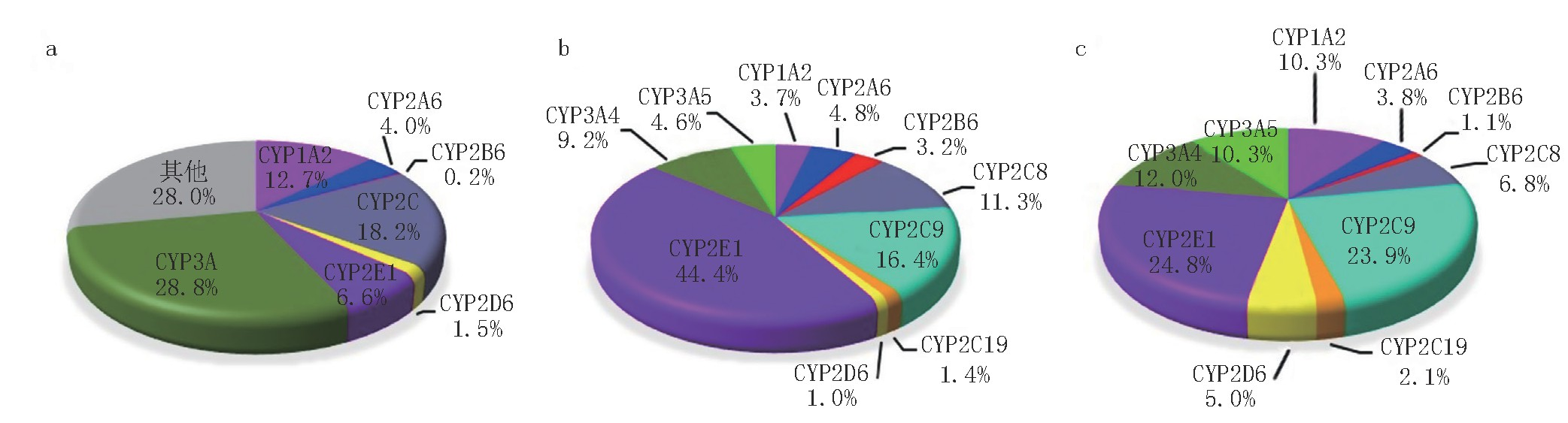
▲ 图源 | 药学进展,人肝CYP450酶含量与活性研究进展
代谢酶介导的DDI研究策略图
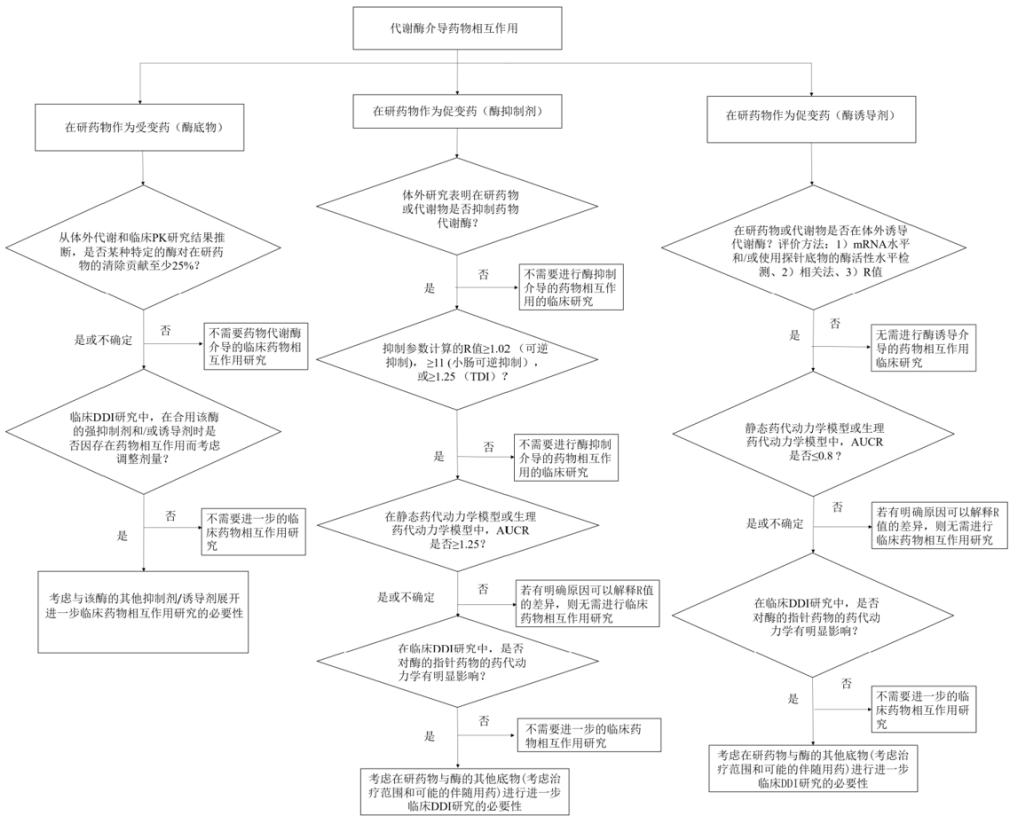
▲ 图源 | CDE《药物相互作用研究技术指导原则(试行)》
DDI的主要研究内容包括但不限于:
1
是否是促变药
在研药物是否可改变其它药物的药代动力学特征;
2
是否是受变药
其它药物是否可改变在研药物的药代动力学特征
3
DDI的程度
评估在研药物药代动力学参数的变化程度
4
DDI的临床意义
评估在研药物DDI的临床意义
5
DDI的预防措施
临床严重DDI的防控策略
二、临床DDI研究的设计与实施要点
ICHM12系统规范了临床DDI研究的类型、设计和实施要求,问答文件对样本量等关键参数提供了具体指导。
2.1 研究类型选择
1️⃣独立与嵌套DDI研究
独立研究以DDI评价为主要目的;嵌套研究作为其他临床试验(如II/III期)的一部分,需预先规划和适当设计。
2️⃣指针药物研究
使用强效指针抑制剂/诱导剂评估最大DDI潜力,结果可外推至相同机制的其他药物。
3️⃣预期合并用药研究
评价与目标人群可能合用药物的DDI,更具临床针对性但其结果可能难以外推至其他药物。
4️⃣鸡尾酒法
同时评价多种酶/转运体的抑制/诱导潜力。
5️⃣生物标志物法
使用内源性底物(如CPI、4β-羟基胆固醇)评估DDI潜力。
2.2 关键设计考虑
1️⃣样本量
通常每组12-20例,变异大或特定目的时需增加。应能可靠评估DDI程度和变异度。
2️⃣剂量选择
促变药:临床最大剂量+最短间隔(评价最大DDI可能);
受变药:线性范围内任选剂量,或最可能观察到DDI的治疗剂量。
3️⃣给药方案
抑制剂:多剂量给药(除非仅吸收环节作用);
诱导剂:7-14天预处理以达到最大诱导;
底物:若无非时间依赖性PK,可单次给药,若其具有时间依赖性药代动力学特征,则底物药物和促变药均应以多次给药的方式进行DDI评价。
4️⃣研究人群
健康受试者(可外推时)或目标患者人群(安全性考虑时)。
5️⃣试验设计
优先选择交叉设计(减少变异);
若半衰期长,可采用平行设计(需更大样本量)。
6️⃣采样与数据分析
采样需覆盖完整AUC(包括半衰期延长情况);
检测原形药+关键代谢产物(若机制复杂);
可稀疏采样促变药以确认暴露水平。
2.3 特定酶/转运体DDI评价注意事项
1️⃣CYP酶底物
首选强指针抑制剂/诱导剂;
若结果阴性,无需进一步研究相同酶的其他促变药;
若阳性,进一步评估中效促变药或使用模型预测。
2️⃣CYP酶抑制剂或诱导剂
底物选择:首选对目标CYP酶活性变化最敏感、特异性高的指针底物进行初始研究。需综合考虑底物的特异性和在研药物的抑制/诱导特征。
研究决策:
阴性结果:若与最敏感底物无相互作用,则无需研究该通路敏感性更低的其他底物。
阳性结果:若发现存在相互作用,则应进一步研究相关的合并用药底物,并评估其临床意义。
复杂情况:若在研药物同时具有抑制和诱导作用,其净效应可能随时间变化(初期抑制为主,后期诱导为主)。研究设计需包含早期和晚期的药代动力学采样点,以捕捉这种动态变化。
3️⃣作为UGT底物的药物
DDI幅度:UGT抑制导致的DDI通常比CYP酶介导的DDI程度更小。
研究决策:是否进行临床DDI研究取决于药物的安全窗和与UGT抑制剂合并使用的可能性。
其他考量:
需注意UGT抑制剂可能影响其他途径,导致复杂机制。
建议同时监测原形药及其葡萄糖醛酸代谢物,以阐明机制并评估活性/毒性代谢物的风险。
可利用UGT基因多态性数据来评估代谢途径的重要性和潜在DDI程度。
需注意CYP3A诱导剂(通过PXR/CAR)也可能诱导UGT,从而影响UGT底物。
4️⃣作为UGT抑制剂的药物
UGT抑制的临床意义通常有限,一般不强制要求进行临床DDI研究。
决策时需综合考虑体外抑制数据和与特定UGT底物合并使用的可能性及该底物的安全性。
5️⃣作为UGT诱导剂的药物
UGT可被PXR/CAR激动剂(如CYP3A诱导剂)诱导,但诱导程度弱于CYP3A。
研究策略:如果一个药物在临床研究中被证实是中效或强效CYP3A诱导剂(使敏感底物AUC降低≥50%),则应考虑其诱导UGT的潜力,并基于与UGT底物合用的可能性和安全窗等因素,决定是否进行专门的UGT DDI研究。
特别注意:某些药物可能同时抑制CYP3A但诱导UGT,需综合判断其净效应。
2.4 结果解读标准
无效应界值
最佳基于暴露-效应关系,默认80-125%一般可接受。
三、案例分享——Dasatinib(Sprycel®)
Dasatinib(达沙替尼)是一种由百时美施贵宝(BMS)开发的酪氨酸激酶抑制剂,用于治疗慢性期、加速期或急变期的慢性髓细胞白血病(CML),以及对既往治疗耐药或不耐受的费城染色体阳性(Ph+)急性淋巴细胞白血病(ALL)。
药物名称:Dasatinib(Sprycel®)
化学名称:N-(2-氯-6-甲基苯基)-2({6-[4-(2-羟基乙基)哌嗪基-1]-2-甲基嘧啶基-4}氨基)-1.3-噻唑-5-酰胺,一水合物
主要成分:达沙替尼
化学结构式:
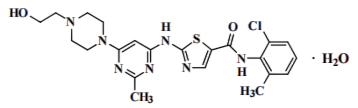
分子式:C22H26ClN7O2S•H2O
分子量:506.02(一水合物)
体外研究表明,达沙替尼是一种时间依赖性的CYP3A4抑制剂,因此可能影响CYP3A4底物的药代动力学。
体内研究如下:
试验设计:
为了研究达沙替尼的抑制潜力,进行了与辛伐他汀(一种CYP3A4底物)的药物相互作用研究。48名健康成年受试者在空腹条件下接受以下两种治疗,每种治疗之间均有7天的洗脱期:
•队列A:辛伐他汀 80mg(单次剂量)
•队列B:辛伐他汀 80mg与达沙替尼100mg
由于达沙替尼是一种时间依赖性的CYP3A4抑制剂,因此达沙替尼的剂量应达到稳态水平因此,本研究的结果可能低估了达沙替尼对CYP3A4的抑制潜力。
结果如下:

▲ 图源 | Dasatinib ablets ,Clinical pharmacology and biopharmaceutics riview(s), FDA.
几何均值比(Cmax)和AUC的90%可信区间表明,达沙替尼的联合使用会增加辛伐他汀的暴露量。当与单次剂量的100毫克达沙替尼联合使用时,辛伐他汀的暴露量增加了20%至23%,Cmax增加约37%。这表明达沙替尼具有CYP3A4抑制潜力,但其抑制作用不足以被视为弱抑制剂。由于该研究的设计并非最优化,达沙替尼的抑制潜力可能比本试验结论更为显著。
说明书中体现为:
达沙替尼与CYP3A4底物同时使用可能会增加CYP3A4底物的暴露。因此,当达沙替尼与具有较窄治疗指数的CYP3A4底物联用时应当谨慎,这些底物包括阿司咪唑、特非那定、西沙必利、匹莫齐特、奎尼丁、苄普地尔或麦角生物碱类(麦角胺、双氢麦角胺)。
更多详情 · 请联系
电话:021-3178-5055
邮箱:yiyao-marketing@weipugroup.com

扫描上方二维码
关注微谱公众号
电话:021-3178-5055
总部:上海市杨浦区国伟路135号9号楼
邮箱:yiyao-marketing@weipugroup.com
本网站中提及的资质、荣誉等相关数据来源:微谱科技集团旗下分子公司及其关联公司;
本网站中提及的各项业务,由拥有相应业务资质的微谱科技集团旗下分子公司及其关联公司承接;其中专利代理业务由上海微略知识产权代理有限公司全权受理。





