
专家解读 | CHO重组表达产品上市申报前遗传稳定性研究要求
2024/12/18

一、什么是“胃pH依赖性药物相互作用”?
要理解这个概念,得先从“胃酸”和“抑制胃酸药物(ARA)”说起。
我们的胃里有胃酸,正常空腹时pH值约1.0-3.0,像个“酸性熔炉”。很多口服药要在这个“熔炉”里溶解、被肠道吸收,才能发挥作用,而抑制胃酸药物(ARA),会改变这个“熔炉”的环境,当胃pH升高,有些药物的溶解度会降低或升高,导致吸收量变多或变少。简单理解就是:抑制胃酸药物(ARA)改变了胃环境,间接影响了其他药物的“吸收效率”。
二、什么药物具有发生pH依赖性DDI的可能性?
1
弱碱性药物常释制剂(DDI风险最高)
特点:在酸性环境(正常胃酸)里溶解度高,容易溶解吸收;一旦胃酸被抑制(pH升高到6.0-6.8),溶解度会骤降,吸收量减少,可能导致药效不足。
弱碱性药物常释制剂与ARA合用的临床DDI风险评估流程如下:
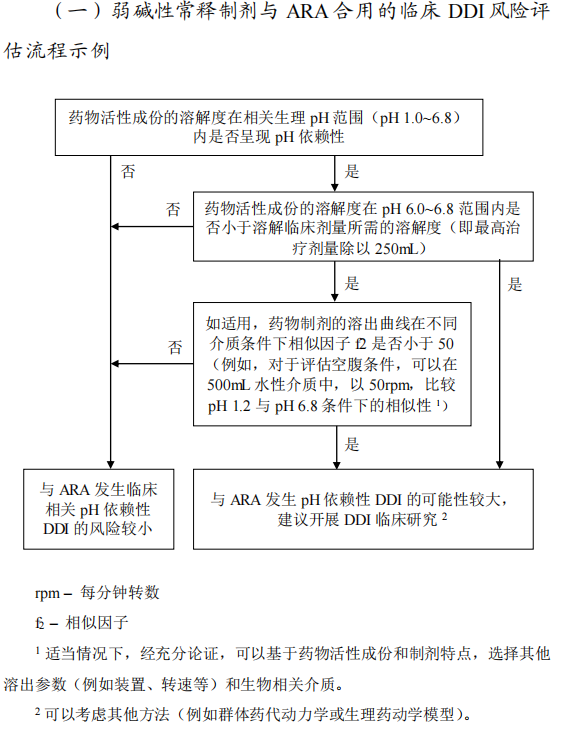
2
弱酸性药物常释制剂(影响较小)
特点:和弱碱性药物相反,弱酸性药物在酸性环境(胃酸)里溶解度低,在抑酸后的碱性环境里溶解度会升高。
但根据国内外研究数据,这类药物的吸收变化通常不大,很少引发严重问题。CDE与FDA的这两份文件均指出其DDI程度通常较小,是否需临床研究取决于药物安全性特征或剂量-暴露安全性关系。
3
调释制剂(特殊情况)
特点:这类药的“释放机制”依赖胃酸环境(比如肠溶片在胃里不溶解,到肠道才溶解)。如果用了ARA,胃pH升高,可能导致药物提前释放(比如肠溶片在胃里就溶解了),不仅可能刺激胃黏膜,还会改变吸收节奏。
仅pH敏感释放机制的缓释或迟释制剂可能发生DDI,需结合制剂释放机制综合考量,均建议特殊情况咨询监管/审核部门。
三、怎么开展pH依赖性的DDI临床研究?
试验关键设计
▶
1、研究人群
优先健康受试者,细胞毒性等安全性风险高的药物,在目标患者人群中开展;需足够样本量以评估DDI程度与变异。
▶
2、研究设计
首选交叉设计(固定序列/随机)以减少个体差异;药物半衰期长时可采用平行组设计。
▶
3、抑制胃酸药物的选择
PPI(首选,“最坏情况”评估):国内建议预用4~7天达药效稳态,FDA建议4~5天,且FDA明确举例40mg埃索美拉唑、20mg雷贝拉唑以达到近最大pH升高效应。
H₂阻断剂:研究药给药前2小时单次/多次给予,因作用持续时间短,可通过错时给药(如研究药在H₂阻断剂前2小时或后10~12小时)降低风险。
抗酸剂:单次与研究药同服,因作用时间短,错时2小时可规避风险。
其他考虑因素:避免选择有其他相互作用机制的ARA,例如奥美拉唑是一种已知的CYP2C19抑制剂,西咪替丁抑制多种CYP酶和转运体(如CYP2D6、CYP3A4、MATE1和MATE2/K)。
▶
4、给药剂量
ARA:采用临床常用最大剂量,以表征最大DDI效。
研究药:采用拟定的最大推荐治疗剂量(更易受pH影响)。
▶
5、制剂处方
优先使用拟上市处方,早期处方需提供外推至拟上市处方的证据。
▶
6、研究药物的给药频次
通常研究药物采用单次给药方式。若多次给药后药物吸收发生改变,或者必须采用患者开展研究且单次给药对需要持续治疗的患者没有获益,则建议考虑采用多次给药方式。
▶
7、进食条件
空腹条件更能反映最大DDI风险(高脂餐可能因刺激胆汁酸分泌低估风险);若研究药需随餐服用,按后期临床试验进食条件开展。
▶
8、样本采集与数据收集
需覆盖药代动力学关键指标(AUC0₋inf/AUC0₋tau、Cmax、Tmax,必要时建议表征Cmin或部分AUC活性代谢产物(影响疗效/安全性)需同步检测。
四、如何运用临床研究的结果?
结果外推:同类别ARA(如不同PPI)在相似pH升高效应下可外推;若ARA有其他相互作用机制,外推需谨慎。
防控策略:
若研究显示临床意义DDI:
PPI:建议避免合用,或开展低剂量PPI的补充研究。
H₂阻断剂:建议避免合用,或通过临床研究验证错时给药方案。
抗酸剂:推荐错时给药(研究药在抗酸剂前2小时或后2小时),必要时验证更短间隔可行性。
弱碱性常释制剂DDI临床研究结果外推示例图如下:
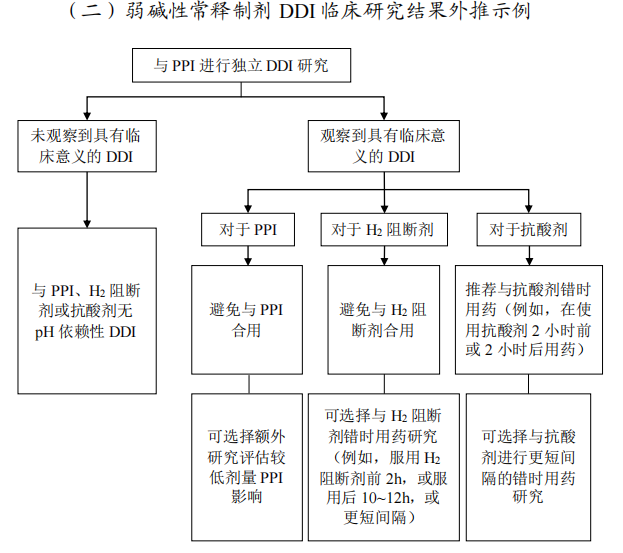
五、能否不开展临床研究?
两份文件均认可非临床评估手段的辅助作用:建模与模拟方法。
1
群体药代动力学(PopPK)
需前瞻性记录给药信息(ARA/研究药剂量、时间、食物),强化吸收相血样采集,按ARA类别(PPI、H₂阻断剂、抗酸剂)作为独立协变量分析,对比持续/偶尔使用ARA患者的药物暴露。
2
生理药代动力学(PBPK)
主要用于弱碱性药物,结合生物相关介质溶出数据提升预测力,辅助临床研究设计;FDA特别强调需咨询审核部门,国内则要求验证模型可靠性。
六、总结
胃酸依赖性药物相互作用不是“所有药都冲突”,若pH依赖性DDI的评估策略和评估方法存在特殊情况,可事先与监管机构沟通。
七、案例分享——Dasatinib(Sprycel®)
Dasatinib(达沙替尼)是一种由百时美施贵宝(BMS)开发的酪氨酸激酶抑制剂,用于治疗慢性期、加速期或急变期的慢性髓细胞白血病(CML),以及对既往治疗耐药或不耐受的费城染色体阳性(Ph+)急性淋巴细胞白血病(ALL)。
药物名称:Dasatinib(Sprycel®)
化学名称:N-(2-氯-6-甲基苯基)-2({6-[4-(2-羟基乙基)哌嗪基-1]-2-甲基嘧啶基-4}氨基)-1.3-噻唑-5-酰胺,一水合物
主要成分:达沙替尼
化学结构式:
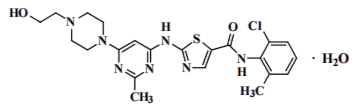
分子式:C22H26ClN7O2S•H2O
分子量:506.02(一水合物)
溶解度:达沙替尼表现出pH依赖的溶解度,在正常生理pH范围内,溶解度随着pH值的增加而降低。
胃pH依赖性DDI研究:
申请人提交了一项关于达沙替尼、抗酸药和H2受体拮抗剂之间相互作用的研究。以下治疗方案以交叉方式施用于24名健康受试者:
▶
1、治疗A
Dasatinib 50mg,每12小时一次(对照组)
▶
2、治疗B
晚上剂量:Dasatinib 50mg,在服用40mg口服法莫替丁前2小时服用
上午剂量:Dasatinib 50mg(在法莫替丁剂量后10小时)
▶
3、治疗C
晚上剂量:Maalox 30mL,在Dasatinib 50mg前2小时服用
上午剂量:Dasatinib 50mg,与Maalox 30mL一起服用
结果如下:
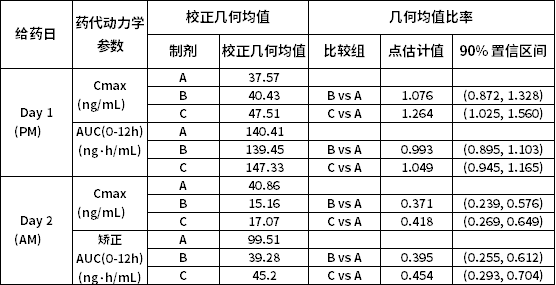
法莫替丁在Dasatinib给药后2小时使用,不会对Dasatinib的AUC产生任何影响。然而,Dasatinib的Cmax在第一天治疗后略有增加(7%)。但是,当Dasatinib在法莫替丁给药后10小时使用(治疗B,第二天)时,会出现显著下降。Dasatinib在法莫替丁给药后10小时使用时,Cmax减少63%,AUC0-12减少60%。
对于治疗C,在服用Maalox后2小时给予Dasatinib并未显著改变AUC(增加4%),但Cmax确实增加约26%,这被认为在临床上并不重要。然而,在第2天(上午剂量)同时服用Dasatinib和Maalox确实导致Dasatinib的Cmax显著下降(58%)和AUC0-12显著降低(54%)。
最终这个DDI研究的结论也相应地呈现在了本品的说明书中:
在单次给药SPRYCEL前2小时给予30mL氢氧化铝/氢氧化镁,与达沙替尼平均AUC无相关变化;然而,Dasatinib的平均Cmax增加了26%。
同时给予30mL氢氧化铝/氢氧化镁与单次剂量SPRYCEL给药,导致达沙替尼平均AUC降低55%,达沙替尼平均Cmax降低58%。
法莫替丁(H2拮抗剂)给药后10小时单次给予SPRYCEL可使Dasatinib的平均AUC降低61%,Dasatinib的平均Cmax降低63%。
国内上市说明书中明确:
在接受本品治疗的患者中,应当考虑使用抗酸药替换H2拮抗剂或质子泵抑制剂。抗酸药可在本品给药前2小时或给药后2小时服用。
更多详情 · 请联系
电话:021-3178-5055
邮箱:yiyao-marketing@weipugroup.com

扫描上方二维码
关注微谱公众号
电话:021-3178-5055
总部:上海市杨浦区国伟路135号9号楼
邮箱:yiyao-marketing@weipugroup.com
本网站中提及的资质、荣誉等相关数据来源:微谱科技集团旗下分子公司及其关联公司;
本网站中提及的各项业务,由拥有相应业务资质的微谱科技集团旗下分子公司及其关联公司承接;其中专利代理业务由上海微略知识产权代理有限公司全权受理。





