
专家解读 | CHO重组表达产品上市申报前遗传稳定性研究要求
2024/12/18


赵清佳
微谱化药事业部毒理学研究专家
中国毒理学会认证毒理学家
赵清佳,微谱化药事业部毒理学研究专家,中国医药工业研究总院药物分析专业硕士,中国毒理学会认证毒理学家,深耕毒物分析技术开发与代谢机制研究领域。她拥有7年毒理学领域专业知识沉淀,含4年国内外药品及化学品毒理学风险评估实战经验,专注于为产品提供从研发到上市的全流程安全性合规解决方案。赵老师精通 ICH S 系列(药品)、OECD 测试指南、ECETOC TRA 及 REACH 等多国法规体系,能精准适配 NMPA、FDA、EMA等不同监管市场的合规要求,拥有丰富的产品申报/核查经验。

当已上市药品或申报品种中发现新的或水平升高的非致突变杂质,且其含量超过ICH Q3A/Q3B规定的界定阈值(QT)时,应如何进行科学、有效、合理的安全性评估?长期以来,行业普遍采用在原料药中“加样”该杂质,然后用此加样批次进行重复给药毒性动物实验。然而,该方法存在科学价值有限及违背3R伦理原则的 “双重困境”。

▲ 《关于非致突变杂质的界定的思考性文件》图源丨EMA官网
EMA《关于非致突变杂质的界定的思考性文件》的核心使命是为行业提供一套可替代传统动物实验、更具科学性和特异性的非临床安全评估策略,以填补ICH Q3A/Q3B在“超阈值新杂质或超限杂质”评估指导上的不足。该文件适用于化学合成原料药和制剂中的非致突变杂质,不适用于先进治疗药品、草药及生物制品。
针对上述问题,该文件构建了一套基于证据权重(Weight of Evidence,WoE)的风险评估决策流程图(见图1),其核心思想是:优先使用非动物方法(体内/体外毒理学数据、代谢物数据、交叉参照(Read-Across)以及NAMs(New approach methodologies)等进行安全性评估,仅在所有替代方法均无法解决安全隐患时,才考虑进行动物实验。
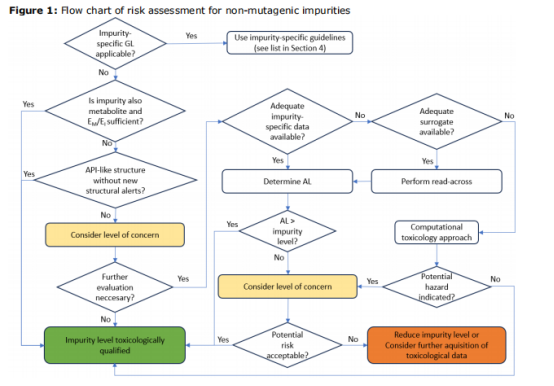
▲ 图1 基于证据权重(WoE)的风险评估决策流程
图源丨《关于非致突变杂质的界定的思考性文件》EMA官网
优先解决措施与建议如下:
1
代谢物法
如果该杂质在动物或人体的(非)临床研究中已被证实为代谢物,且其暴露水平(EM)与作为杂质摄入的预期暴露水平(EI)之比(EM/EI)足够高(通常≥1,对高风险代谢物需≥10),则可直接认定其安全性已被覆盖,无需额外数据。
2
Read-Across评估法
如果杂质本身不能查询出足够可靠的体外/体内毒性数据,可以根据其化学结构、物理化学性质、药代动力学等表征结果,采用单一替代或分组方法应用相似物的体外/体内毒性数据对该杂质进行安全性评估。
对于化学结构、理化性质与原料药高度相似,且未引入新的“警示结构”或“毒性基团”的杂质(如某些二聚体、水解/氧化产物),可认为其毒性特征已被原料药的毒理学研究充分覆盖。这与ICH M7中 “Class 4”杂质(与无致突变性的原料药具有相同警示结构)的逻辑一脉相承。
3
“关注水平”评估法
这是一个多因素综合判断过程,其关键考虑因素包括:
①最大每日摄入量:参考毒理学关注阈值(TTC) 概念。文件指出,虽然1.5 µg/天(源自ICH M7)是低关注起点,但1 mg/天常被用作一个重要的参考点:
摄入量低于TTC阈值则说明需关注度低;
介于TTC与1 mg/天之间则需个案评估;
高于1 mg/天则关注度显著升高。
②治疗时长:短于终生(LTL)暴露可允许更高的日摄入量,这与ICH M7中基于累积风险调整LTL限度的原则一致。
③患者人群:儿科、肝肾功能不全、孕妇等敏感人群需特别考虑。
④给药途径与临床适应症:严重或危及生命的疾病可容忍更高的风险。
此外,该文件详述了可接受水平(AL)计算方法论。如果关注水平评估认为需要进一步风险评估,则推荐计算AL。AL法借鉴了ICH Q3C(残留溶剂)和Q3D(元素杂质)中允许日暴露量(PDE) 的计算原理,但更具有灵活性。
关键评估起点(PoD):推荐优先使用基准剂量下限(BMDL),其次为未观察到的不良反应水平(NOAEL)等。
应用不确定因子(AFs):系统性地考虑了种属差异(AF1)、个体差异(AF2)、研究时长(AF3)、毒性严重程度(AF4)、LOAEL使用(AF5)、给药途径生物利用度差异(AF6)以及Read-Across评估的不确定性(AF7)。
产品特异性:可接受限度的计算公式为:AL(μg/d) = [PoD(mg/kg/d) × 50 kg × 1000] / (AF1 × AF2 × AF3 × AF4 × AF5 × AF6 × AF7)。
AL计算时需考虑特定药品使用背景,非通用限值。
4
NAMs
NAMs为当传统毒理学数据(如文献、交叉参照)不足时,用于支持非致突变杂质风险评估的补充性工具。其目的为在无需进行新的体内动物研究的情况下,填补特定的数据缺口,为杂质的危害识别或剂量反应评估提供证据。
①计算毒理学,应用符合OECD验证原则的(Q)SAR模型,识别警示结构、预测杂质的毒性效力;这与ICH M7要求使用两种互补的(Q)SAR方法进行致突变性预测的理念相呼应,但本文件将其扩展至非致突变性终点(如肝毒性、肾毒性等)。
②体外方法,使用细胞系、原代细胞、类器官或微生理系统等建立的体外测试模型,用于评估特定毒性终点、获取生物学数据;
③不良结局路径(AOPs),整合多源数据,构建从分子起始事件到不良反应的链条,用于定性或定量风险评估。
④危害表征与定量风险评估,若NAMs数据表明杂质对相关靶器官缺乏危害,此信息可作为证据权重的一部分,支持“风险可接受”的结论。若NAMs数据提示潜在危害,则评估者必须结合暴露水平、药代动力学考量及所有其他可用数据,进行综合判断,以论证在拟定质量标准限度下的暴露是否不存在安全风险。
最后,仅当上述所有方法均无法支持杂质在拟定标准下的安全性时,才考虑进行动物实验。文件对此提出了设计建议:必须使用纯度>95%的分离杂质,而非“加样”原料药,以确保观察到的效应确由杂质引起;推荐采用至少4个剂量组,以支持BMDL计算;纳入毒代动力学(TK)评估;遵循OECD测试指南等。
微谱拥有丰富的药品/化学品毒理评估项目经验、具备中国和美国毒理学家资质认证的技术团队。已为百余家客户提供了药物活性物质HBEL/PDE评估报告,助力客户通过药品共线生产清洁验证现场核查。微谱团队拥有近5000种非活性物质/杂质毒性数据库及Case Ultra基毒预测评估软件,致力于帮助客户解决杂质研究、香精成分小分子醛类物质等研究难题,助力企业顺利通过申报。
更多详情 · 请联系
电话:021-3178-5055
邮箱:yiyao-marketing@weipugroup.com

扫描上方二维码
关注微谱公众号
电话:021-3178-5055
总部:上海市杨浦区国伟路135号9号楼
邮箱:yiyao-marketing@weipugroup.com
本网站中提及的资质、荣誉等相关数据来源:微谱科技集团旗下分子公司及其关联公司;
本网站中提及的各项业务,由拥有相应业务资质的微谱科技集团旗下分子公司及其关联公司承接;其中专利代理业务由上海微略知识产权代理有限公司全权受理。





